

Case Study: Spice up the Food and Beverage Industry with AI-Engineered Fermentation Enzyme
Download

EnzymoGenius™ is a leading provider of docking-based protein-ligand interaction analysis services for drug discovery. Our comprehensive approach integrates state-of-the-art computational methodologies to unravel intricate molecular interactions crucial for drug design and development.
Docking-based protein-ligand interaction analysis plays a pivotal role in drug discovery, employing computational algorithms to predict the binding affinity between a protein and a potential drug molecule. This method facilitates the identification of novel drug candidates by simulating the docking process, elucidating the energetically favorable conformations of ligands within the protein's binding site. Recent research progress in this domain focuses on refining docking algorithms to enhance accuracy and efficiency, incorporating advanced scoring functions and considering receptor flexibility. Additionally, advancements in parallel computing and machine learning techniques contribute to the robustness of predictions. These developments propel the application of docking-based approaches in the virtual screening of vast chemical libraries, streamlining the drug discovery pipeline by prioritizing compounds with high binding affinity and therapeutic potential.
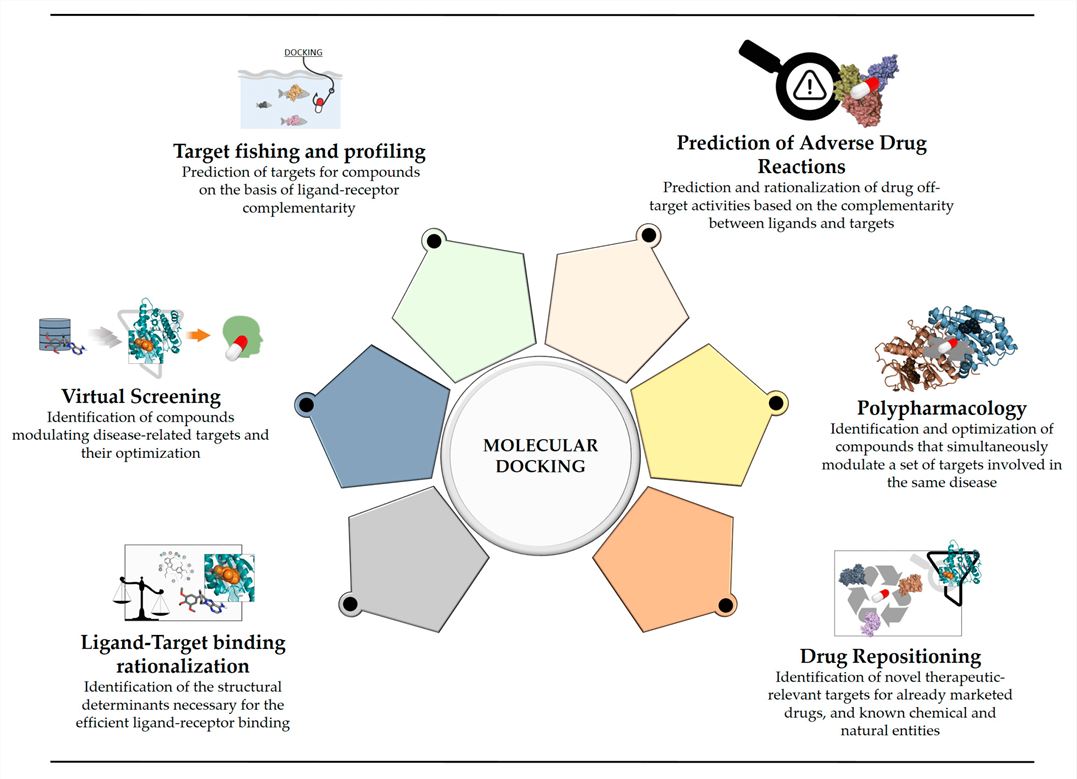 Fig. 1 Main applications of molecular docking in current drug discovery. (Pinzi L, et al., 2019)
Fig. 1 Main applications of molecular docking in current drug discovery. (Pinzi L, et al., 2019)
EnzymoGenius™ stands at the forefront of computational biology, offering unparalleled expertise in docking-based protein-ligand interaction analysis. Our services encompass molecular docking, binding energy calculation, structural analysis, virtual screening, and ADME/T properties prediction. What sets us apart is our commitment to employing advanced algorithms, providing customized solutions, and integrating structural analyses for a comprehensive understanding of molecular interactions. For innovative solutions in drug discovery, please don't hesitate to contact us.
Reference
Please take a moment to fill out the form.